
Her geçen gün, özellikle teknolojinin hızlı ilerlemesi ile, insan vücudu ve çalışması hakkında yeni şeyler öğreniyoruz. Ama hala kendini gölgeler arasında saklayan gizemler varlığını sürdürmekte. Tıp dünyası, insan vücudunun çeşitli yönleriyle ilgili birçok farklı ve nadir durumu içinde barındırır. Bazıları o kadar ender görülür ki, genellikle literatürde yeterince dikkat çekemezler. Bunlar “Hocam, gerçek hayatta bunları nerede kullanacağız?” sorusunun muhatabı durumlardır. Ancak bu nadir sendromlar hem tıp alanında, hem de genel olarak insan vücudunun karmaşıklığı hakkında ilginç bilgiler sunabilirler. Bu yazımızda nadir ve ilginç sendromlara odaklanacağız.
Elbette, ender görülen sendromlar hakkında bir yazı yazmak oldukça ilginç bir süreçti. Bu konuda araştırma yapmak ve yazıya dökmek benim için oldukça keyifli oldu. Umarım, sizler de bu yazıyı okurken aynı derecede ilgi ve zevk duyarsınız.
Giriş
Bir sendrom, doğrudan nedeni her zaman anlaşılmamış olsa da, belirli bir durumu işaret eden semptomlar ve fiziksel bulguların tanınabilir bir kompleksi olarak tanımlanır.1 Sendrom kelimesi, tek başına belirli bir hastalığı tanımlamak için kullanılmak yerine, genellikle bir dizi semptomun bir araya gelmesiyle karakterize edilen, belirli bir durumu ifade etmek için kullanılır. Tıbbi literatürde, farklı hastalıkların veya durumların sendromlar olarak tanımlandığı birçok örnek bulunmaktadır. Bu terim genellikle belirli semptom grubunun veya belirtilerin karakteristik kombinasyonunun tanımlanması için kullanılır. En bilinen sendromlar için Down Sendromu (Trizomi 21), Asperger Sendromu (Otizm spektrum bozukluğunun bir türü), Klinefelter sendromu (XXY) örnek olarak verilebilir.
Bu yazımızda ise daha az popüler olan, hatta bazıları için “acaba efsane mi?” diye bilim insanlarının üzerinde düşündüğü ender görülen ve duyulan sendromlardan bahsedeceğiz.
Alice Harikalar Diyarında Sendromu (AHDS)

Lewis Carroll’ın (Charles Lutwidge Dodgson) 1865 tarihli Alice Harikalar Diyarında adlı eserinde, kahramanımız Alice vücudunun boyutlarının değişmesinden esinlenen İngiliz psikiyatrist John Todd (1914–1987), 1955 yılında Alice Harikalar Diyarında Sendromu’nu (AHDS) hastanın kendi bedeni veya uzaydaki konumunun boyut, kitle veya şeklindeki bozulmaları içeren, kişinin kendi deneyimlediği paroksismal bir beden-imajı yanılsamaları olarak tanımladı.2
Somestetik algı bozukluklarını içeren vakalardan farklılıklar içeren bu sendrom genellikle çocuklarda izlendi. Bildirilen nedenler arasında enfeksiyon (özellikle Epstein Barr virüsü)3, migren, epilepsi, depresyon ve toksik ve ateşli deliryum bulunmaktadır.2

-_Plate_I.jpg){kind=link}
Joris Hoefnagel, Animalia Rationalia et Insecta (Ignis)-, National Gallery of Art Washington
Kurtadam (Werewolf) Sendromu (Hipertrikoz)
Hypertrichosis, vücutta aşırı derecede saç büyümesini ifade eder. Bu, saçın tüm yüzü ve vücudu kaplayabileceği veya küçük yamalarda meydana gelebileceği nadir bir durumdur. Hem erkekleri hem de kadınları etkileyebilir. Edinsel hipertrikoz, nevoid hipertrikoz, doğumsal hipertrikoz lanuginosa, doğumsal hipertrikoz terminalis ve hirsutizm olmak üzere birkaç türü bulunmaktadır. Bu hastalar, vellus, lanugo veya terminal tip saçlara sahip olabilir.4
Belirli bir etiyoloji bulunmamakla birlikte, porfiria kutanea tarda, androjenik steroidler, liken simplex veya kötü beslenme ile ilişkilendirilebilir. Ayrıca, minoksidil, fenitoin ve siklosporin gibi ilaçlar anormal saç büyümesiyle ilişkilendirilmiştir. Belirli bir tedavi olmadığı için, sadece epilasyon, tıraş, elektroliz veya lazer cerrahisi yoluyla yönetilebilir.4
Stendhal Sendromu

Stendhal sendromu, güzel bir sanat eserinin yanında bulunulduğunda ortaya çıkan farklı semptomların hem fiziksel hem de zihinsel, birleşimi ile karakterize edilen bir durumdur.5 Sendrom, Dr. Graziella Magherini tarafından 1989 yılında ortaya tanımlanmış ve 19. yüzyıl romantik yazarı Marie-Henri Beyle’in (genellikle takma adıyla “Stendhal” olarak bilinir) onuruna bu ad verilmiştir. Dr. Graziella Magherini, Floransa’daki Santa Maria Nuova Hastanesi’nde görülen 106 yabancı (İtalyan olmayan) hastanın durumunu değerlendirmiştir. Bu hastalar, aynı şehirdeki Basilica di Santa Croce’yi ziyaret ettikten sonra Stendhal’ın yazılarıyla uyumlu klinik belirtiler sergilemiştir; bu nedenle durum aynı zamanda “Floransa sendromu” olarak da adlandırılır.6
1986’da Japon psikiyatrist Hiroaki Ota tarafından tanımlanan benzer bir durum vardır. Paris’i ziyaret eden birkaç kişi, baş dönmesi, taşikardi, kalp çarpıntısı, nefes darlığı gibi semptomlarla birlikte, görsel ve işitsel halüsinasyonlar, paranoid takipçi sanrıları ve kişilik bozuklukları gibi psikiyatrik semptomlar göstermiştir. Dr. Ota’nın bulgularına göre, bu klinik tablo, seyahat eden kişinin kendi kültüründen oldukça farklı bir kültür ve yaşam tarzıyla karşılaşmasının yarattığı önemli etkiyle olabilir.7
Farklı bir dünya ile karşılaşma ile ilişkili başka bir hastalık da Dr. Bar-El ve meslektaşları tarafından gözlemlenmiştir. “Kudüs sendromu” olarak bilinen bu durum, Stendhal sendromunun oldukça benzer özelliklerine sahiptir ve aynı zamanda üç dünyanın en çok takip edilen, etkili ve güçlü dinleri tarafından genel olarak “kutsal toprak” olarak kabul edilen bir şehri ziyaret ettiğinde mesihçi fikirlerin ve büyüklük sanrılarının bulunmasıyla dikkat çeker. Venezia, Roma ve İstanbul gibi diğer şehirler ve simgeler, çeşitli yazarlar tarafından bu estetik sendromlarla ilişkilendirilmiştir.8
Yabancı El Sendromu (Alien Hand)
Yabancı El Sendromu, corpus callosum ve yardımcı motor alanın lezyonlarıyla ilişkili nadir bir durumdur. Etyolojide nörocerrahi, tümör, anevrizma ve nadiren inme vardır. 9Genellikle beyin cerrahisi sonrası görülür. Sendrom, bir kişinin kendi kontrolü dışında olan el veya uzvunu kontrol edememe durumudur. Bu uzuv, genellikle kişinin kontrolü dışında bir şekilde hareket eder, kişiye dokunur veya istenmeyen eylemler gerçekleştirebilir.10 Bir el çekmecenin kapağını kapatabilirken, diğeri açabilir. Yabancı el işbirliği yapmaz ve muhtemelen komutları takip etmekte başarısız olabilir.
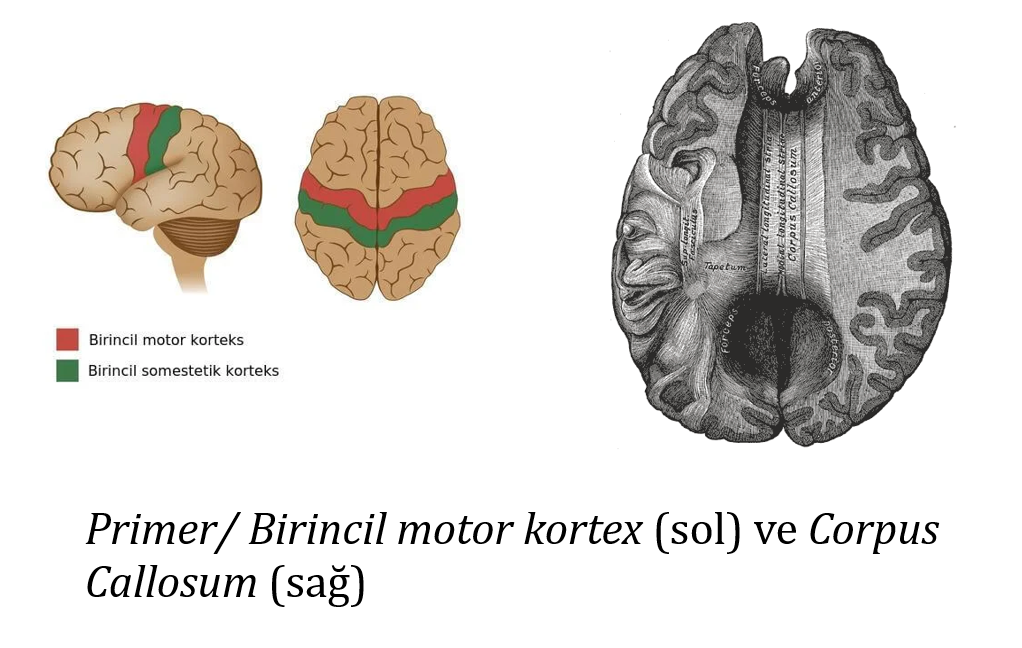
Bu durum beynin iki yarım küresinin ayrılmasından kaynaklanır. Bu, corpus callosum ve yardımcı motor alanın lezyonlarıyla ilişkili nadir bir durumdur. Taramalarda, bu sendromu olan kişilerin birincil/primer motor kortekste izole aktiviteye sahip olduğu gösterilmektedir. Bu durumun, hareket planlama ve kontrolünden sorumlu olan parietal korteksteki hasardan kaynaklandığı düşünülmektedir. Bu bölgenin hasarıyla, koordinasyonsuz planlama ve spontan hareket meydana gelir.11 Yani el, kendi beyni varmış gibi bağımsız hareket eder.
Progeria Sendromu (Hutchinson-Gilford Progeria Sendromu -HGPS)
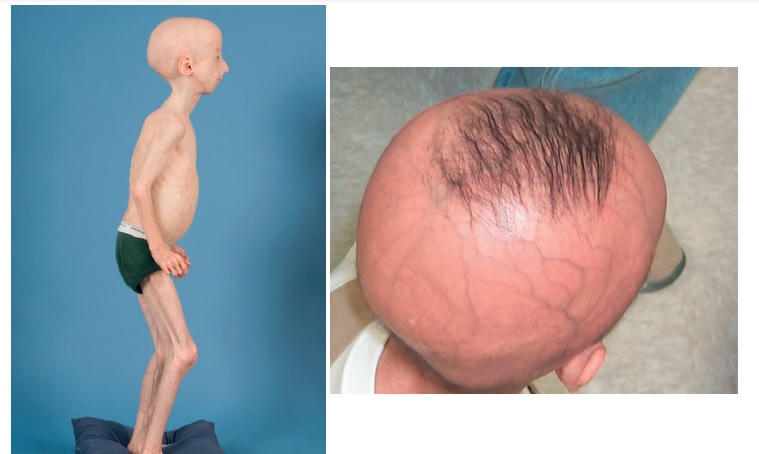
Progeria, Hutchinson-Gilford Progeria Sendromu (HGPS) olarak da bilinen progeroid sendromlardan biridir. Tahmini genel prevalansı 18 milyonda birdir.12 Sendrom; hücresel ve organizma düzeyinde yaşlanma sürecine ilişkin bakış açısı kazanmamıza yardımcı olabilecek fenotipler gösteren, son derece nadir, tek tip ölümcül bir “erken yaşlanma” hastalığıdır. Progeria ile yaşlanma arasındaki en önemli genetik bağlantı, her replikasyon döngüsünde telomer uçlarının kısalmasıdır. Hastaların genellikle son derece kısa telomerleri olduğu gözlemlenmiştir. Son çalışmalarda, HGPS olan bireylerin %90’ında LMNA geninde de-novo nokta mutasyonları olduğu bulunmuştur. LMNA, lamin A ve C’yi kodlar ve A tipi laminler çekirdek zarında önemli bir yapısal fonksiyona sahiptir. HGPS mutasyonunun en yaygın türü, kodon 608’de (G608G) yer almaktadır.13
Bu hastalık doğumda teşhis edilemez, ancak 2 yaşından sonra belirgin semptomlar gözlemlenebilir. Erken çocukluk dönemindeki ilk belirtiler, büyüme ve dermatolojik bulgulara dayanır. HGPS’li çocukların başlıca morbiditesi ve mortalitesi aterosklerotik kardiyovasküler hastalık ve inmelerden kaynaklanmakta olup, ölüm genellikle ortalama 14,6 yaşında meydana gelmektedir. Genetik geçiş beklenmez, çünkü hastalar genellikle üreme çağından önce ölürler.14
HGPS’li hastalarda, hem büyük hem de küçük damar hastalığı mevcuttur ve inmeler genellikle klinik olarak sessizdir. Çoklu sistemlerde erken yaşlanma bulunmasına rağmen; HGPS’li çocukların bilişsel bozulma yaşamadıkları gözlemlenmiştir. Sınırlı otopsi materyaline dayanarak, demans veya Alzheimer tipi değişikliklere dair patolojik kanıt bulunmamaktadır. Önemli bir şekilde, nükleer bozulmalar hipokampal nöronlarda önemli değişikliklere neden olmamıştır.14
Rett Sendromu

1954 yılında, Avusturya’nın Viyana şehrinde pediatri uzmanı Dr. Andreas Rett, aynı tekrarlayan el yıkama hareketlerini yapan iki kız çocuğunu gözlemledi. Onların klinik ve gelişimsel geçmişlerini karşılaştırdı ve çok benzer olduklarını keşfetti. Dr. Rett, benzer bir davranış gösteren altı kız çocuğu daha buldu ve bu kızların filmlerini çekerek Avrupa’nın çeşitli yerlerinde benzer belirtileri olan diğer çocukları aramak için seyahat etti. Bulgularını 1966 yılında birkaç Alman tıp dergisinde yayımladı. Dr. Bengt Hagberg ve arkadaşları; 1982’de bazı kız çocuklarında benzer belirtileri fark etti ve bu sendrom hakkında daha ayrıntılı bir makale yazdı. Yazarlar sendromun öncüsü Dr. Andreas Rett adını onurlandırarak sendroma Rett Sendromu adını verdiler.15
Rett sendromu (RTT), X-kromozom bağlantılı bir gen olan metil-CpG bağlayıcı protein 2 (MECP2) genindeki mutasyonlar tarafından oluşturulan bir nörolojik bozukluktur. Bu gen, yaygın olarak ifade edilen bir transkripsiyonel düzenleyici olarak işlev görür. Genellikle kızlarda görülen bu sendrom, normal gelişimden sonra gerileyen bir süreçle birlikte motor ve bilişsel yeteneklerde kayba sebep olan bir nörogelişimsel bozukluktur.16
RTT, açık bir şekilde normal erken gelişimi takiben iletişimsel ve ince motor becerilerin gerilemesi ile karakterizedir. Epilepsi, ciddi bilişsel bozukluklar ve otonom ve motor işlev bozuklukları gibi eşlik eden durumlar bulunmaktadır.17 RTT genellikle kız çocuklarında görülür. Tipik Rett sendromu için tanısal kriterler, bir gerileme dönemini, bunu takip eden iyileşme veya stabilizasyonu ve dört ana kriterin tümünün (amaçlı el becerilerinin kaybı, konuşma dilinin kaybı, yürüme anormallikleri ve stereotipik el hareketleri) karşılanmasını gerektirir. Ocak 2023’te yaynılanan Rett sendromunun küresel yaygınlığı: Sistematik inceleme ve meta-analiz’nde prevalansı 100.000 kadında 5 ila 10 vaka olarak tahmin edilmektedir.18
Keşfinden bu yana önemli bilimsel ilerlemelere rağmen, MECP2 mutasyonlarının RTT belirtilerine nasıl yol açtığı mekanizması büyük ölçüde bilinmemektedir. Sonuç olarak, hastalar için mevcut tedavi seçenekleri şu anda sınırlıdır ve semptomları hafifletmeye odaklanmıştır.16 Yaklaşık 60 klinik deneme ve gen terapisi vaadi olmasına rağmen, henüz tedavi belirtilere yöneliktir ve bir tedavi ortaya çıkmamıştır.17
Moebius (Möbius) Sendromu
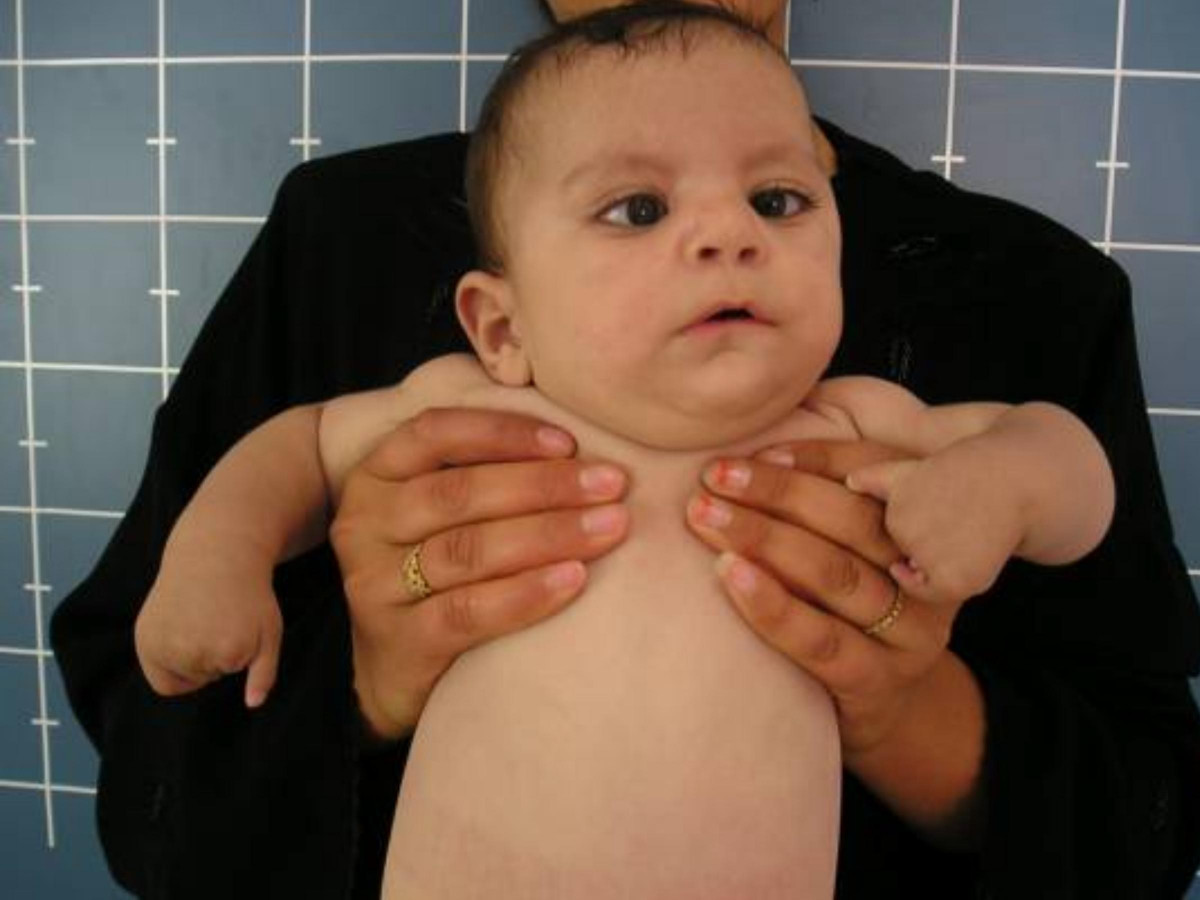
Moebius (Möbius) sendromu (MBS); tek taraflı, bilateral simetrik ya da asimetrik Fasiyal (VII) ve Abdüsens (VI) sinir felçleri ile karakterize nadir bir doğuştan gelişen kranial sinir bozukluğudur. Genetik ve intrauterin çevresel etkileşimlere bağlı olarak beyincik damar bozuklukları, gelişimine katkıda bulunabilir.19 Sendrom, ilk kez 1888 yılında tanımlayan Alman nörolog Paul Julius Möbius’un adını taşımaktadır.20
Farklı ağız-yüz anomalileri ile karşımıza çıkabilir. Tanı tamamen klinik esaslara dayanmasına rağmen, bazı karakteristik özellikler, beyin görüntülerinde mevcuttur. Bulgular, embriyogenez sırasında beyincik damarlarında intrauterin vasküler bozulma teorisinin en yaygın etiyoloji olduğunu, genetik hipotezin bunu takip ettiğini öne sürmektedir. İntrauterin çevresel etkileşimler, potansiyel risk faktörleri olarak gösterilmiştir. Fasiyal ve Abdüsens sinir felçleri en yaygın sunum özellikleridir. Ancak, alt kranial sinirlerin (IX, X, XI, XII) klinik belirtileri, ortopedik anomaliler ve zihinsel eksikliklerle birlikte de görülebilir olabilir. Tanı, tanımlanmış az sayıda tanı kriteri ile klinik temellere dayanır. Beyincik ve beyin sapını içeren karakteristik radyolojik bulgular, görüntüleme çalışmalarında gözlemlenebilir. Radyolojik bulgulara rağmen, Moebius sendromu klinik olarak tanımlanır.21
Henüz kesin tedavisi olmayan sendromda, kişiye özel yönetim stratejileri önemlidir. Kesin tedavi seçenekleri olmamasına rağmen, destekleyici bakım sağlamak için multidisipliner bir yaklaşım benimsenir. Tedavi edilemez olmasına rağmen, kişiye özel rehabilitasyon önlemleri ile fiziksel ve psikolojik eksiklikler yönetilebilir; ancak standart yönergelerin belirlenmesi gereklidir.21
Fil Adam / Proteus Sendromu
1862 ‘de normal sağlıklı bir bebek olarak dünyaya gelen Joseph Merrick’in büyüdükçe cilt ve kemiklerinde ciddi anormallikler gelişti. Çoklu ekzositozlar, büyümüş bir kafa ve cildin altında gevşek, sarkık, papillomatöz ve verrüköz olan kalınlaşma ortaya çıktı. Sağ kolunda ve bir bacağında asimetri vardı. Herhangi bir cafe-au-lait lekesi belirgin değildi ve ayağının alçı dökümü, taban yüzeyinde serebriform bağ dokusu nevüsünü gösteriyordu. Kendisine “fil adam” lakabı takılarak yıllarca kafes hayvanı muamelesi gördü. Hayatı, gösterilerden biri sırasında Londra’da çalışan bir doktorun onu fark etmesi ile değişti. Hastaneye yatırılan Joseph Merrick 1890’da hayatını kaybetti. Hayatı çeşitlitli eserler konu olan Joseph Merrick, 1986 da Proteus Sendromu tanısını aldı.23
Proteus sendromu terimi, 1983 yılında iskelet, hamartomatöz ve diğer mezodermal anormalliklerin bir bozukluğunu tanımlamak için kullanıldı. Sendrom, adını “Çok Biçimli” anlamına gelen Yunan tanrısı Proteus’tan almıştır.24

Proteus sendromu, aktive edici bir AKT1 mutasyonu (c.49G>A, p.Glu17Lys) tarafından oluşturulur. Bu mozaik bozukluğun sebep olduğu birçok değişken özellik mümkündür. Orantısız, asimetrik ve şekli bozan aşırı büyüme; diğer bozukluklarda gözlenenden farklı kemik anormallikleri; yüksek oranda kolajen içeren karakteristik serebriform bağ dokusu nevüsü; erken yaşlarda akantoz ve hiperkeratozdan oluşan epidermal nevüsler; kapiller, venöz veya lenfatik tiplerde vasküler anormallikler; lipomalar, lipohipoplazi, yağlı büyüme ve yerel yağ birikimlerini içeren düzensiz yağ dokusu; büllöz akciğer değişiklikleri; belirli neoplazmalar; zihinsel engel ve/veya nöbetlerle ilişkilendirilen yüz görünümü, ve/veya beyin anormallikleri; ve erken ölüme neden olan derin ven trombozu bu mozaik gen bozukluğunun yol açtığı durumlardır.23
Hastaların yarısından fazlasında bulunan ana klinik bulgular arasında; hemihypertrofi, makrodaktili, ekzositozlar, epidermal nevüsler, ayak tabanı veya avuç içini içeren karakteristik serebriform kitleler, çeşitli deri altı kitleler ve skolyoz bulunmaktadır. Deri altı kitlelerin histolojik incelemesi, çeşitli yağlı, hamartomatöz ve anjiomatöz tümörleri tanımlamıştır.24
Son söz
Nadir görülen sendromlar genellikle sadece az sayıda insanın yaşadığı, ancak insan vücudunun ve psikolojisinin karmaşıklığı hakkında önemli bilgiler içeren durumlardır. Bu sendromlar, daha fazla araştırma ve anlayış gerektiren ilginç tıbbi ve psikolojik fenomenlerdir. Onları anlamak, hem tıp alanında ilerlememize hem de insan doğasını daha iyi kavramamıza yardımcı olabilir. Bu sendromlar oldukça nadir görülen durumlardır ve çoğu zaman spesifik semptomları ve belirtileri nedeniyle tanımlanırlar. Her biri genellikle kendi benzersiz semptomları, belirtileri ve tedavi yöntemleriyle öne çıkar. Bu sendromlar üzerine yapılan araştırmalar, daha iyi anlaşılmasını ve potansiyel tedavi seçeneklerinin geliştirilmesini sağlayabilir.
Kaynaklar
- 1.Calvo F, Karras B, Phillips R, Kimball A, Wolf F. Diagnoses, syndromes, and diseases: a knowledge representation problem. AMIA Annu Symp Proc. 2003;2003:802. https://www.ncbi.nlm.nih.gov/pubmed/14728307
- 2.Lanska D, Lanska J. The Alice-in-Wonderland Syndrome. Front Neurol Neurosci. 2018;42:142-150. doi:10.1159/000475722
- 3.O’Toole P, Modestino E. Alice in Wonderland Syndrome: A real life version of Lewis Carroll’s novel. Brain Dev. 2017;39(6):470-474. doi:10.1016/j.braindev.2017.01.004
- 4.Joshi T, Kuchewar V. A rare clinical image of hypertrichosis (Werewolf syndrome). Pan Afr Med J. 2022;43:99. doi:10.11604/pamj.2022.43.99.37150
- 5.Innocenti C, Fioravanti G, Spiti R, Faravelli C. [The Stendhal syndrome between psychoanalysis and neuroscience]. Riv Psichiatr. 2014;49(2):61-66. doi:10.1708/1461.16139
- 6.Picard D, Robinson M. Emotion in Motion: Tourism, Affect and Transformation. Routledge; 2012.
- 7.Nicholson T, Pariante C, McLoughlin D. Stendhal syndrome: a case of cultural overload. BMJ Case Rep. 2009;2009. doi:10.1136/bcr.06.2008.0317
- 8.Palacios-Sánchez L, Botero-Meneses JS, Pachón RP, Hernández LBP, Triana-Melo J del P, Ramírez-Rodríguez S. Stendhal syndrome: a clinical and historical overview. Arq Neuro-Psiquiatr. Published online February 2018:120-123. doi:10.1590/0004-282×20170189
- 9.Park Y, Kim C, Kim M, Jeong H, Jung H. Alien hand syndrome in stroke – case report & neurophysiologic study -. Ann Rehabil Med. 2012;36(4):556-560. doi:10.5535/arm.2012.36.4.556
- 10.Lawson M, Lobsien E, Leinisch E, Lobsien D. Alien hand syndrome in ruptured aneurysms: case report and review of the literature. Neuroradiology. 2022;64(10):2091-2094. doi:10.1007/s00234-022-03025-5
- 11.Panikkath R, Panikkath D, Mojumder D, Nugent K. The alien hand syndrome. Proc (Bayl Univ Med Cent). 2014;27(3):219-220. doi:10.1080/08998280.2014.11929115
- 12.Gordon CM, Gordon LB, Snyder BD, et al. Hutchinson‐gilford progeria is a skeletal dysplasia. J of Bone & Mineral Res. Published online June 21, 2011:1670-1679. doi:10.1002/jbmr.392
- 13.Ahmed M, Ikram S, Bibi N, Mir A. Hutchinson-Gilford Progeria Syndrome: A Premature Aging Disease. Mol Neurobiol. 2018;55(5):4417-4427. doi:10.1007/s12035-017-0610-7
- 14.Ullrich N, Gordon L. Hutchinson-Gilford progeria syndrome. Handb Clin Neurol. 2015;132:249-264. doi:10.1016/B978-0-444-62702-5.00018-4
- 15.IRSF . About Rett Syndrome . International Rett Syndrome Foundation. Published 2014. Accessed 2023. https://www.rettsyndrome.org
- 16.Kyle S, Vashi N, Justice M. Rett syndrome: a neurological disorder with metabolic components. Open Biol. 2018;8(2). doi:10.1098/rsob.170216
- 17.Coorey B, Haase F, Ellaway C, Clarke A, Lisowski L, Gold W. Gene Editing and Rett Syndrome: Does It Make the Cut? CRISPR J. 2022;5(4):490-499. doi:10.1089/crispr.2022.0020
- 18.Petriti U, Dudman D, Scosyrev E, Lopez-Leon S. Global prevalence of Rett syndrome: systematic review and meta-analysis. Syst Rev. 2023;12(1):5. doi:10.1186/s13643-023-02169-6
- 19.Yaqoob A, Dar W, Raina A, et al. Moebius Syndrome. Ann Indian Acad Neurol. 2021;24(6):929. doi:10.4103/aian.AIAN_182_21
- 20.Möbius PJ. Ueber angeborene doppelseitige Abducens-Facialis-Lahmung. Münchener Medizinische Wochenschrift. 1888;35:91-94.
- 21.Zaidi S, Syed I, Tahir U, Noor T, Choudhry M. Moebius Syndrome: What We Know So Far. Cureus. 2023;15(2):e35187. doi:10.7759/cureus.35187
- 22.Kaissi AA, Grill F, Safi H, Ghachem MB, Chehida FB, Klaushofer K. Craniocervical junction malformation in a child with Oromandibular-limb hypogenesis-Möbius syndrome. Orphanet J Rare Dis. Published online January 8, 2007. doi:10.1186/1750-1172-2-2
- 23.Cohen M. Proteus syndrome review: molecular, clinical, and pathologic features. Clin Genet. 2014;85(2):111-119. doi:10.1111/cge.12266
- 24.Samlaska C, Levin S, James W, Benson P, Walker J, Perlik P. Proteus syndrome. Arch Dermatol. 1989;125(8):1109-1114. https://www.ncbi.nlm.nih.gov/pubmed/2667470